Naba
2015-09-21 08:00:23 UTC
Dear Gromacs users and Developers,
I have performed dihedral PCA for 4 extracellular loops of a transmembrane
protein after successfully finishing 100 ns of simulation at 300 and 310 K.
I obtained figures for each loops for two different temperatures as in this
link: Loading Image...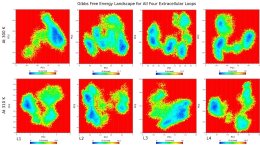 .
.
As we can see in that figure that, the loop L1 has got only one minimum at
both temperatures, whereas, L2 and L3 have got 4 minima at 300 K. I have
also calculated the amount of frames in percentages at their respective
minima. It seems that L2 and L3 have got more number of frames at their
minima when combined in comparison to L1. Now my question is:
Can we say that L2 and L3 are tend to be more stable at 300K than that of
L1 at the both temperatures?
Please help.
Thanks in advance.
Regards
Nabajyoti Goswami
College of Veterinary Science
Khanapara, Guwahati 781022
Assam, India.
I have performed dihedral PCA for 4 extracellular loops of a transmembrane
protein after successfully finishing 100 ns of simulation at 300 and 310 K.
I obtained figures for each loops for two different temperatures as in this
link: Loading Image...
 .
.As we can see in that figure that, the loop L1 has got only one minimum at
both temperatures, whereas, L2 and L3 have got 4 minima at 300 K. I have
also calculated the amount of frames in percentages at their respective
minima. It seems that L2 and L3 have got more number of frames at their
minima when combined in comparison to L1. Now my question is:
Can we say that L2 and L3 are tend to be more stable at 300K than that of
L1 at the both temperatures?
Please help.
Thanks in advance.
Regards
Nabajyoti Goswami
College of Veterinary Science
Khanapara, Guwahati 781022
Assam, India.
--
Gromacs Users mailing list
* Please search the archive at http://www.gromacs.org/Support/Mailing_Lists/GMX-Users_List before posting!
* Can't post? Read http://www.gromacs.org/Support/Mailing_Lists
* For (un)subscribe requests visit
https://maillist.sys.kth.se/mailman/listinfo/gromacs.org_gmx-users or send a mail to gmx-users-***@gromacs.org.
Gromacs Users mailing list
* Please search the archive at http://www.gromacs.org/Support/Mailing_Lists/GMX-Users_List before posting!
* Can't post? Read http://www.gromacs.org/Support/Mailing_Lists
* For (un)subscribe requests visit
https://maillist.sys.kth.se/mailman/listinfo/gromacs.org_gmx-users or send a mail to gmx-users-***@gromacs.org.